
DNA서열을 아미노산 서열로 변환하기(with python)
1주차 스터디를 성황리에 마치고 2주차에 접어들었다.
1주차는 파이썬 기본기를 다뤄서 크게 어려울 것이 없었는데
2주차부터는 난이도가 다소 높아질 것 같다.
앞으로의 포로그램은 주피터 노트북에서 실행할 예정이다.
그럼 … 시작 !!
DNA 프로젝트
인터넷에서 제공하는 DNA 서열을 다음의 표와 같이 아미노산 서열로 변환한다.
| DNA | ATA | TGG | CAA |
|---|---|---|---|
| 아미노산 | I | W | Q |
과제 진행 순서
- DNA와 프로틴 시퀀스 데이터를 수동 다운로드
- 파이썬으로 DNA 데이터를 열고 가공하기
- DNA를 아미노산으로 변환하는 알고리즘
- 변환한 아미노산 서열과 사이트에서 다운로드한 것이 일치하는지를 비교
1. DNA와 프로틴 시퀀스 데이터를 수동 다운로드
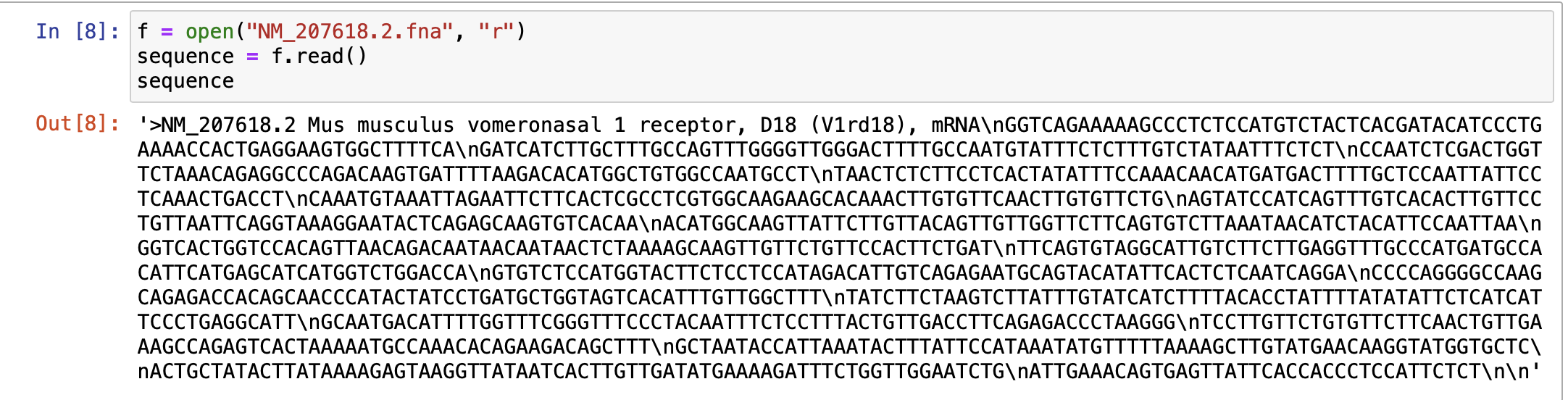
NCBI에서 DNA시퀀스 파일을 다운로드 했다.
파이썬의 open() 명령어를 이용해 다운로드 한 파일을 읽기 전용으로 연 후 출력했다.
f = open("NM_207618.2.fna", "r")
sequence = f.read()
sequence
소스코드와 출력 결과
2. 파이썬으로 DNA 데이터를 열고 가공하기
파일을 열어보면 첫 번째 줄에 염기서열을 설명하는 내용이 있다.
데이터를 분석하는 데 불필요하므로 삭제한다.
with open() 구문을 사용하면 해당 블록을 벗어났을 때 자동으로 파일을 닫아준다.
파일을 읽기 전용으로 열고, splitlines() 메소드를 이용해 줄 단위로 읽어들인다.
splitlines()는 여러 줄의 문자열을 줄바꿈(‘\n’)을 기준으로 나누며,
각 줄의 내용을 요소로 갖는 리스트를 생성한다.
이 때, 매개변수가 True 라면 ‘\n’도 포함해서 리스트를 만든다. (False라면 줄바꿈 문자는 삭제)
소스코드를 작성해보자!
with open("NM_207618.2.fna", "r") as inf: #with 로 열면 블럭이 끝날 때 자동으로 파일 닫아줌
data = inf.read().splitlines(True) #한 줄씩 '\n'포함한 리스트로 저장
with open('dna1.txt', 'w') as outf: # 쓰기 모드로 열기
outf.writelines(data[1:]) # data에 저장된 리스트의 두 번째 요소부터 'dna1.txt' 파일에 쓰기
f = open("dna1.txt", "r") #'dna1.txt' 파일을 읽기모드로 열기
sequence = f.read() # 열어둔 파일을 읽어와서 sequence 라는 변수에 저장
sequence #sequence를 출력해 파일 내용 확인
출력 결과를 보면 다음과 같다.
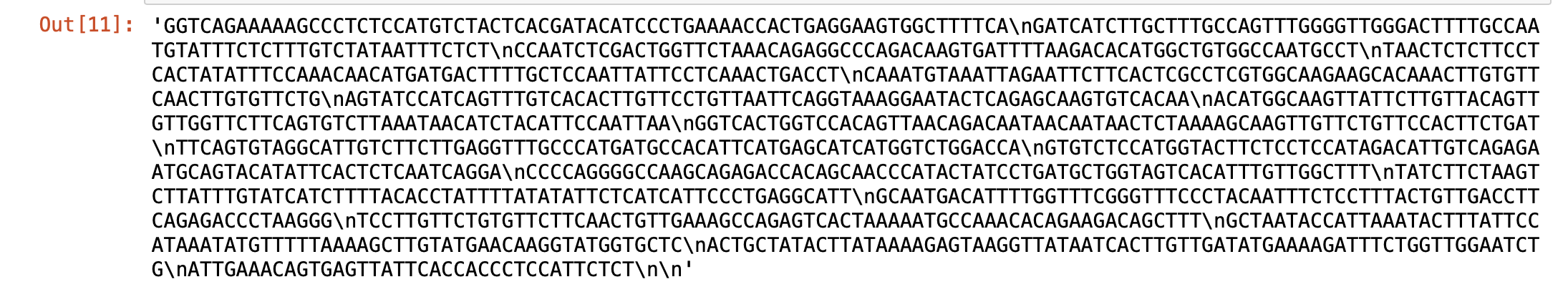
결과에는 ‘\n’ 문자가 포함되어 있다.
그러나 데이터 분석에는 필요하지 않으므로 replace()메소드를 이용해 삭제한다.
엔터키(Carriage return)나 혹시 모를 공란도 역시 삭제한다!

결과를 보면 깔끔히 삭제된 것을 볼 수 있다.
3. DNA를 아미노산으로 변환하는 알고리즘
DNA에 대한 아미노산의 정보를 이용해 dna.txt의 내용을 아미노산 서열로 변환하는 작업이 필요하다.
genetic_code = {
'ATA':'I', 'ATC':'I', 'ATT':'I', 'ATG':'M',
'ACA':'T', 'ACC':'T', 'ACG':'T', 'ACT':'T',
'AAC':'N', 'AAT':'N', 'AAA':'K', 'AAG':'K',
'AGC':'S', 'AGT':'S', 'AGA':'R', 'AGG':'R',
'CTA':'L', 'CTC':'L', 'CTG':'L', 'CTT':'L',
'CCA':'P', 'CCC':'P', 'CCG':'P', 'CCT':'P',
'CAC':'H', 'CAT':'H', 'CAA':'Q', 'CAG':'Q',
'CGA':'R', 'CGC':'R', 'CGG':'R', 'CGT':'R',
'GTA':'V', 'GTC':'V', 'GTG':'V', 'GTT':'V',
'GCA':'A', 'GCC':'A', 'GCG':'A', 'GCT':'A',
'GAC':'D', 'GAT':'D', 'GAA':'E', 'GAG':'E',
'GGA':'G', 'GGC':'G', 'GGG':'G', 'GGT':'G',
'TCA':'S', 'TCC':'S', 'TCG':'S', 'TCT':'S',
'TTC':'F', 'TTT':'F', 'TTA':'L', 'TTG':'L',
'TAC':'Y', 'TAT':'Y', 'TAA':'_', 'TAG':'_' ,
'TGC':'C', 'TGT':'C', 'TGA':'_', 'TGG':'W',
}
print(genetic_code['ATA'])
앞에서 연습한 내용을 바탕으로 다운로드 한 데이터를 가공하는 과정에서 공란, 개행문자, 엔터키를 파일에서 삭제한 후 새로운 파일에 저장하는 함수를 작성했다.
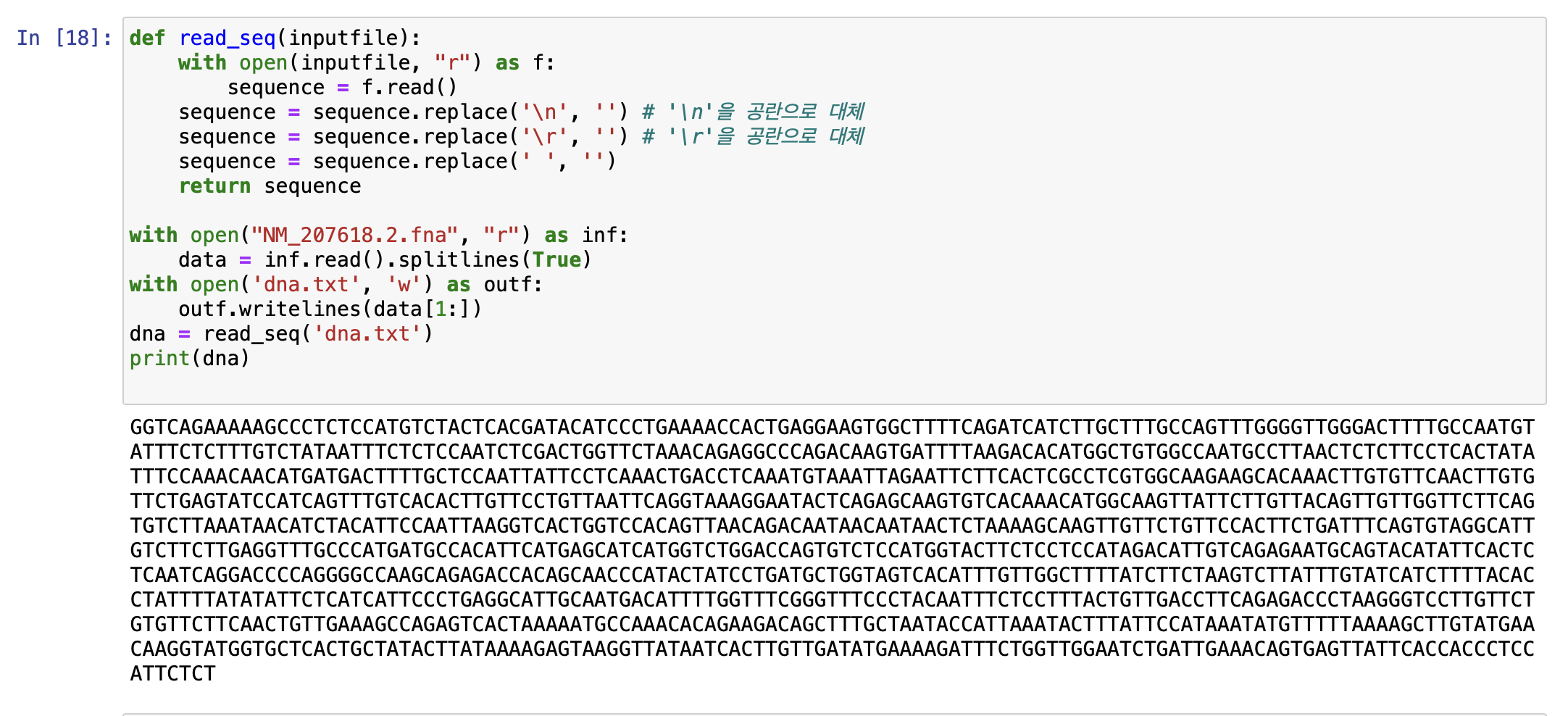
이제 dna.txt 파일에 저장된 DNA 정보를 아미노산 시퀀스로 변환하는 코드를 작성한다.
DNA코드를 3개씩 끊어서 1개의 아미노산 코드로 변환한다.
그런데 NCBI 사이트에서 다운로드한 파일의 일부분만 아미노산의 DNA이므로 어느 부분이 여기에 해당하는지 조사해야 한다. 파일을 다운로드 한 페이지에서 찾아보면, 21..938 이라고 되어있는데, 이게 유전자의 코딩 시퀀스의 시작과 끝을 나타내는 위치다. 우리는 0에서부터 시작하는 배열을 사용하기 때문에 20~937 까지를 사용하면 된다.
(참고: 리스트의 슬라이싱 기능을 활용해 sequence에 저장된 내용을 3개씩 가져올 수 있다. “sequence[0:3]”)
def convert(seq): #DNA시퀀스를 아미노산 시퀀스로 변환
genetic_code = {
'ATA':'I', 'ATC':'I', 'ATT':'I', 'ATG':'M',
'ACA':'T', 'ACC':'T', 'ACG':'T', 'ACT':'T',
'AAC':'N', 'AAT':'N', 'AAA':'K', 'AAG':'K',
'AGC':'S', 'AGT':'S', 'AGA':'R', 'AGG':'R',
'CTA':'L', 'CTC':'L', 'CTG':'L', 'CTT':'L',
'CCA':'P', 'CCC':'P', 'CCG':'P', 'CCT':'P',
'CAC':'H', 'CAT':'H', 'CAA':'Q', 'CAG':'Q',
'CGA':'R', 'CGC':'R', 'CGG':'R', 'CGT':'R',
'GTA':'V', 'GTC':'V', 'GTG':'V', 'GTT':'V',
'GCA':'A', 'GCC':'A', 'GCG':'A', 'GCT':'A',
'GAC':'D', 'GAT':'D', 'GAA':'E', 'GAG':'E',
'GGA':'G', 'GGC':'G', 'GGG':'G', 'GGT':'G',
'TCA':'S', 'TCC':'S', 'TCG':'S', 'TCT':'S',
'TTC':'F', 'TTT':'F', 'TTA':'L', 'TTG':'L',
'TAC':'Y', 'TAT':'Y', 'TAA':'_', 'TAG':'_' ,
'TGC':'C', 'TGT':'C', 'TGA':'_', 'TGG':'W',
}
protein = ""
if len(seq) % 3 == 0: # 데이터의 길이가 3의 배수이면 아래를 실행
for i in range(0, len(seq), 3):
codon = seq[i : i+3]
protein += genetic_code[codon]
return protein
print(convert(dna[20:938]))
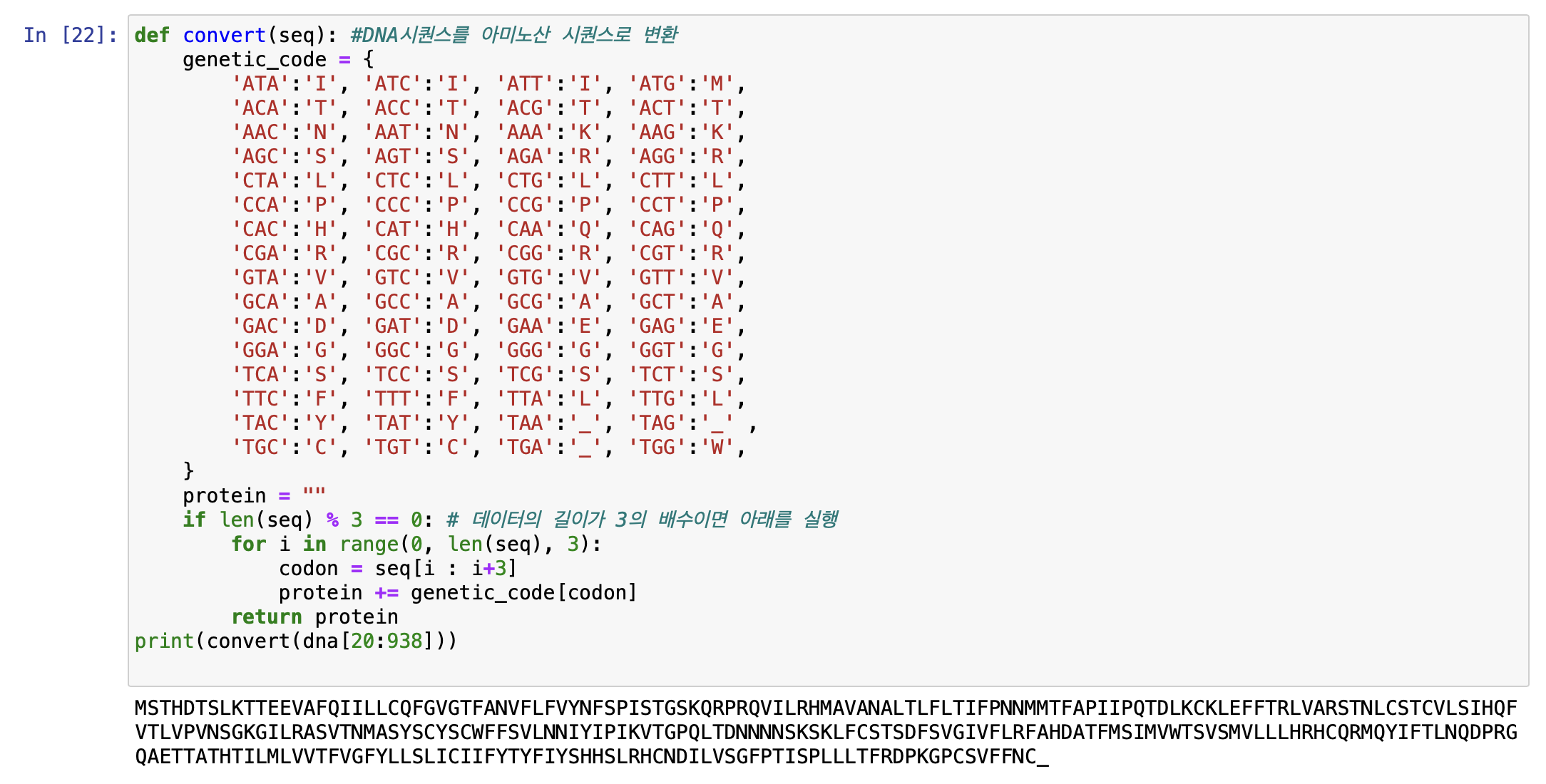
위 소스코드의 출력 결과를 보면 ‘c’ 다음에 ‘_’ 문자가 출력되었다.
이는 EOF와 같은 종결 문자를 포함하기 때문이다.
그래서 마지막 3개 문자는 제외한다 !
print(convert(dna[20:935]))
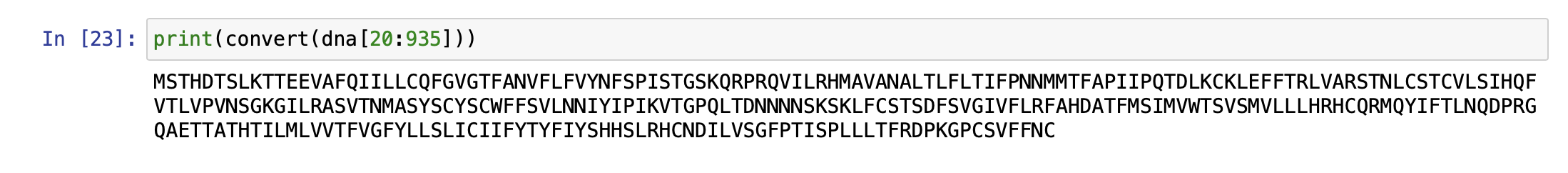
그럼 이렇게 올바른 결과를 얻을 수 있다.
4. 변환한 아미노산 서열과 사이트에서 다운로드한 것이 일치하는지를 비교
지금까지 변환한 아미노산의 결과가 실제 결과와 동일한지 NCBI 사이트에서 확인해보면, 정확하다 !
이렇게 chapter8은 마무리하고 다음 포스팅에서는 chapter9로 넘어가보겠다.